
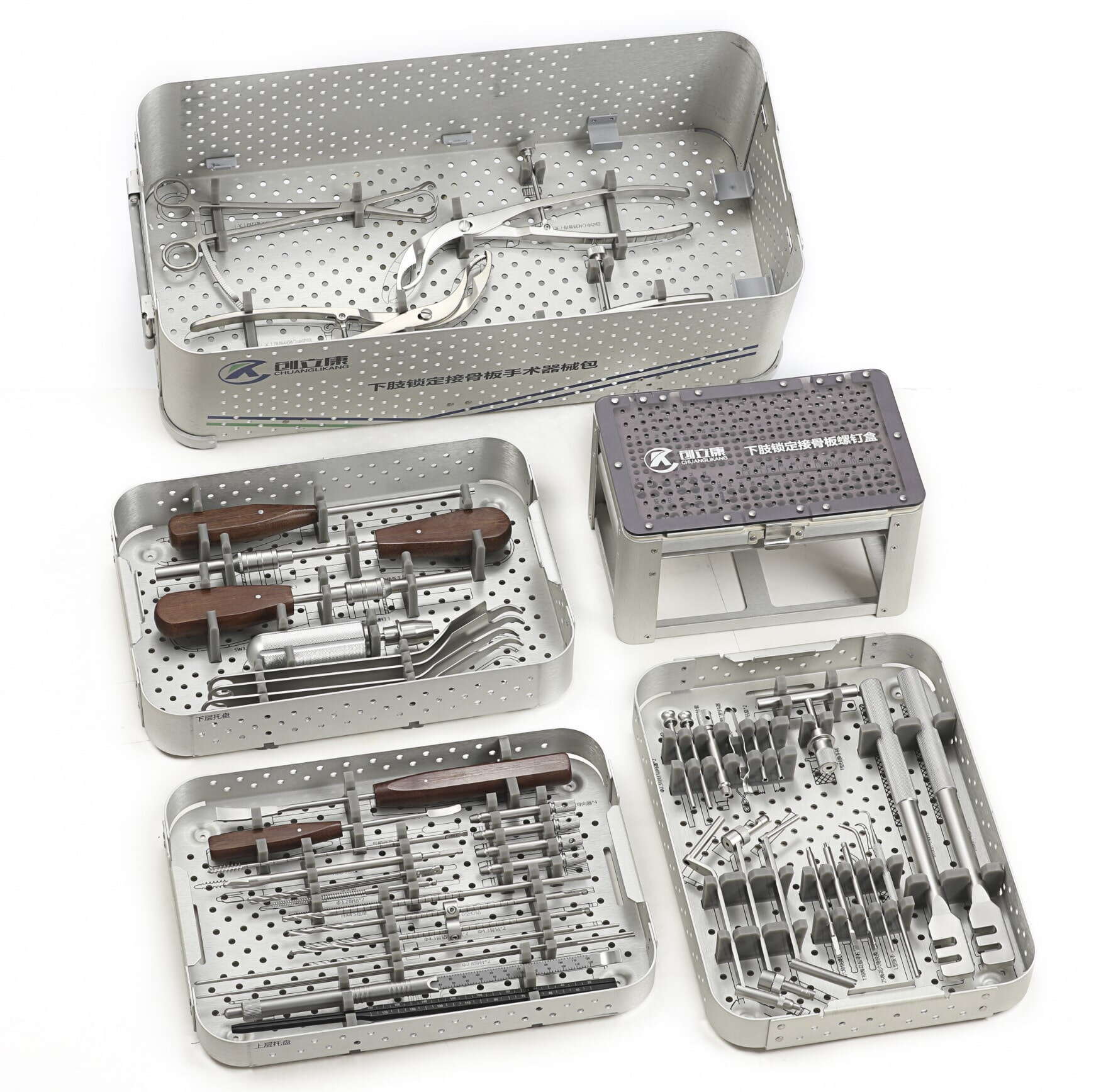
Den globala hälso- och sjukvårdsindustrin är kraftigt beroende av precisionsillkonstruktion, särskilt när det gäller tillverkning av livräddande medicintekniska produkter. En fabrik för ortopediska trauma-instrument verkar under stränga regleringsramar för att säkerställa patientsäkerhet och kirurgisk framgång. Dessa specialiserade tillverkningsanläggningar måste följa omfattande kvalitetskontrollåtgärder som täcker allt från materialval till slutlig produktestning. Komplexiteten hos ortopediska trauma-instrument kräver att tillverkare implementerar omfattande kvalitetsledningssystem som uppfyller internationella standarder samtidigt som de bibehåller kostnadseffektiva produktionsprocesser.
ISO 13485 Kvalitetsstyrning för medicintekniska produkter
Grunden för excellens inom tillverkning av medicintekniska produkter
ISO 13485 utgör grundläggande standard för alla anständiga fabriker som tillverkar ortopediska traumainstrument. Detta omfattande kvalitetsledningssystem tar särskilt hänsyn till de unika kraven inom tillverkning av medicintekniska produkter och säkerställer att varje tillverkningsaspekt uppfyller strikta regelverkskrav. Standarden omfattar designkontroller, riskhantering och processer för kontinuerlig förbättring, vilka är nödvändiga för att bibehålla konsekvent produktkvalitet.
Genomförandet av ISO 13485 kräver systematisk dokumentation av alla tillverkningsprocesser, från initial designidé till leverans av det färdiga produkten. En fabrik för ortopediska traumainstrument måste etablera tydliga procedurer för leverantörsqualificering, granskning av inkommande material samt validering av processer. Dessa krav säkerställer att varje komponent som används i tillverkningen av traumainstrument uppfyller förutbestämda specifikationer och bidrar till den totala säkerheten och effektiviteten hos den färdiga produkten.
Dokumentations- och spårbarhetskrav
Dokumentationskraven enligt ISO 13485 sträcker sig långt bortom enkel arkivföring. Varje fabrik för ortopediska trauma-instrument måste upprätthålla omfattande spårbarhetsregister som spårar material från källan till levereringen av det färdiga produkten. Detta inkluderar detaljerade partiurapporter, kalibreringsintyg för mätutrustning och valideringsstudier som visar processens konsekvens över tid.
Spårbarhet blir särskilt kritiskt vid hantering av implanterbara traumaprodukter, där patientsäkerheten beror på förmågan att snabbt identifiera och återkalla potentiellt defekta produkter. Moderna tillverkningsanläggningar använder sofistikerade databassystem för att underhålla dessa register och säkerställa snabba åtgärdsförmåga vid kvalitetsproblem eller regulatoriska efterfrågningar.
FDA 21 CFR Part 820 Compliance Framework
God tillverkningspraxis krav
FDA:s regel 21 CFR del 820, allmänt känd som Quality System Regulation, fastställer obligatoriska goda tillverkningspraktiker för produktion av medicintekniska produkter i USA. Alla fabriker som tillverkar ortopediska trauma-instrument och som söker godkännande eller tillstånd från FDA måste visa fullständig efterlevnad av dessa omfattande krav. Regeln täcker alla aspekter av produktionsprocessen, från designkontroll och inköp till produktions- och processkontroller.
Designkontroller enligt del 820 kräver att tillverkare etablerar systematiska metoder för att översätta användarnas behov till detaljerade produktspecifikationer. Detta inkluderar genomförande av designgranskningar, verifiering och validering samt upprätthållande av designhistorikfiler. För traumainstrument är dessa kontroller särskilt viktiga med tanke på de kirurgiska tillämpningarnas kritiska karaktär och behovet av exakt funktionalitet i nödsituationer.
Produktions- och processkontroller
Produktionskontroller som krävs enligt del 820 säkerställer att varje fabrik för ortopediska traumainstrument upprätthåller konsekventa tillverkningsprocesser. Dessa kontroller inkluderar skriftliga förfaranden för produktionsmetoder, utrustningsunderhåll och miljöövervakning. Temperatur, fuktighet och renhetsnivåer måste kontinuerligt övervakas och dokumenteras för att förhindra förorening eller nedbrytning av känsliga material.
Processvalidering utgör en annan viktig komponent i efterlevnad av del 820. Tillverkare måste kunna visa att deras produktionsprocesser konsekvent producerar enheter som uppfyller förutbestämda specifikationer. Detta innebär genomförande av installationskvalificering, driftskvalificering och prestandakvalificering för all kritisk tillverkningsutrustning och alla processer.
Materialstandarder och biokompatibilitetstestning
Val av medicinskt gradmaterial
Urvalet av lämpliga material utgör grunden för tillverkning av kvalitetsinstrument för traumavård. En fabrik för ortopediska trauma-instrument måste använda material som uppfyller specifika medicinska kvalitetskrav, inklusive krav på biokompatibilitet, korrosionsbeständighet och mekaniska egenskaper. Rostfria stålsorter som 316LVM och titanlegeringar används ofta på grund av deras utmärkta biokompatibilitetsprofil och mekaniska styrkeegenskaper.
Materialcertifieringsdokumentation måste följa varje leverans av råmaterial och innehålla detaljerad analys av kemisk sammansättning samt verifiering av mekaniska egenskaper. Dessa intyg säkerställer att material uppfyller fastställda specifikationer innan de går in i tillverkningsprocessen. Regelbundna inkommande kontrollförfaranden verifierar att mottagna material överensstämmer med den medföljande certifieringsinformationen.
Protokoll för validering av biokompatibilitet
Biokompatibilitetstest enligt ISO 10993-standarder säkerställer att traumatiska instrument är säkra vid kontakt med patienter. En fabrik för ortopediska trauma-instrument måste genomföra omfattande biologiska utvärderingsstudier, inklusive cytotoxicitetstest, sensibiliseringsstudier och bedömningar av systemisk toxicitet. Dessa tester utvärderar potentiella negativa biologiska reaktioner som kan uppstå under klinisk användning.
Den biologiska utvärderingsprocessen inleds med en noggrann riskbedömning som tar hänsyn till art och varaktighet av patientkontakt. För trauma-instrument som kommer i tillfällig kontakt med vävnad finns specifika testprotokoll för att utvärdera akut toxicitet och lokal vävnadsreaktion. Tillverkare måste bibehålla detaljerade register över alla resultat från biokompatibilitetstester som en del av sina regleringsmässiga ansökningspaket.
Validering av sterilisering och förpackningsstandarder
Terminala steriliseringsprocesser
Effektiv sterilisering utgör en kritisk kvalitetsstandard för alla fabriker som tillverkar ortopediska traumainstrument. De vanligaste steriliseringsmetoderna inkluderar ångsterilisering, etenoxidsterilisering och gammastrålningssterilisering. Varje metod kräver specifika valideringsprotokoll för att visa sterilitetssäkerhetsnivåer på minst 10^-6, vilket innebär att sannolikheten för att en levande mikroorganism överlever steriliseringsprocessen är en på en miljon.
Validering av ångsterilisering innebär genomförande av studier med biologiska indikatorer med Geobacillus stearothermophilus-sporer, som är mycket motståndskraftiga mot fuktig värme. Dessa studier måste visa konsekvent uppnådda sterilitetssäkerhetsnivåer i alla delar av steriliseringslasten. Temperaturkartläggningsstudier säkerställer jämn värdefördelning i hela steriliseringskammaren.
Krav på steril förpackning
Förpackningssystem för sterila traumainstrument måste bibehålla sterilitet fram till användningsögonblicket samtidigt som de ger tillräcklig skydd under transport och lagring. En fabrik för ortopediska traumainstrument måste validera förpackningsmaterial och förseglingsprocesser enligt ASTM- och ISO-standarder. Detta inkluderar genomförande av accelererade åldrandestudier för att visa förpackningens integritet över produktens avsedda hållbarhetstid.
Sterila barriärsystem använder vanligtvis medicinska papper, icke-vävda material eller plastfolier som tillåter sterilantgenomträngning samtidigt som de förhindrar mikrobiell intrång. Förseglingsprocesser för förpackningar kräver validering för att säkerställa konsekvent seglhållfasthet och integritet. Visuella inspektionsförfaranden verifierar att alla förpackningar uppfyller fastställda acceptanskriterier innan slutgiltig släppning.
Miljökontroll och renrumskrav
Krav på klassificering av renrum
Tillverkningsmiljöer inom en fabrik för ortopediska traumainstrument måste uppfylla specifika renhetskrav för att förhindra kontaminering av medicinska instrument. ISO 14644-standarden definierar klassificering av renrum baserat på den högsta tillåtna koncentrationen av luftburna partiklar. De flesta tillverkningsprocesser för traumainstrument kräver klass 100 000 (ISO 8) eller bättre miljöer för montering och förpackningsoperationer.
Program för miljöövervakning bedömer kontinuerligt luftkvalitet, ytrengöring och personalkyra inom tillverkningsområden. Partikelräknare mäter nivån av luftburen förorening, medan ytprovtagning verifierar effektiviteten i rengörings- och desinficeringsförfaranden. Dessa övervakningsaktiviteter genererar data som visar på fortsatt effektiv miljökontroll.
Personals utbildning och klädprocedurer
Mänskliga operatörer utgör en betydande potentiell källa till förorening vid tillverkning av medicintekniska produkter. En fabrik för ortopediska trauma-instrument måste genomföra omfattande utbildningsprogram för personal som omfattar korrekta klädningförfaranden, handhygienprotokoll och beteenderekommendationer i kontrollerade miljöer. Regelbundna utbildningsutvärderingar säkerställer att all personal bibehåller nödvändiga kompetensnivåer.
Klädningförfaranden innebär vanligtvis flera lager av skyddskläder, inklusive hårnät, ansiktsmasker, sterila handskar och helkroppsdräkter. In- och utgångsförfaranden för renrum måste noggrant arrangeras för att förhindra överföring av föroreningar. Luftduschar och klävande mattor hjälper att ta bort lösa partiklar från personal och utrustning innan inträde i kontrollerade områden.
Vanliga frågor
Vilka är de främsta regulatoriska standarder som styr tillverkning av ortopediska trauma-instrument
De viktigaste regleringsstandarderna inkluderar ISO 13485 för kvalitetsledningssystem, FDA 21 CFR Part 820 för god tillverkningspraxis, ISO 10993 för biokompatibilitetstestning och ISO 14644 för renrumsmiljöer. Dessutom måste en fabrik för ortopediska traumainstrument följa ISO 11137 för steriliseringsvalidering samt olika ASTM-standarder för materialens egenskaper och testmetoder.
Hur påverkar materialval kvaliteten på traumainstrument
Materialval påverkar direkt instrumentets prestanda, hållbarhet och patientsäkerhet. En fabrik för ortopediska traumainstrument måste välja material med lämpliga mekaniska egenskaper, korrosionsbeständighet och biokompatibilitetsegenskaper. Vanliga material är 316LVM rostfritt stål och titanlegeringar, vilka erbjuder utmärkta styrka-till-vikt-förhållanden och beprövade biokompatibilitetsprofiler för kirurgiska tillämpningar.
Vilken roll spelar steriliseringsvalidering i tillverkningsstandarder
Steriliseringsvalidering säkerställer att alla trauma-instrument uppnår nödvändiga sterilitetssäkerhetsnivåer innan klinisk användning. En fabrik för ortopediska trauma-instrument måste genomföra omfattande valideringsstudier som visar att steriliseringsprocesser konsekvent eliminerar levande mikroorganismer. Detta inkluderar studier med biologiska indikatorer, temperaturkartläggning och pågående övervakningsprogram som verifierar processens effektivitet över tid.
Varför är miljökontroller viktiga i produktionen av trauma-instrument
Miljökontroller förhindrar kontamination som kan kompromettera instrumentens sterilitet eller funktionalitet. Tillverkningsområden måste uppfylla specifika renhetskrav enligt ISO 14644, med kontinuerlig övervakning av luftburna partiklar, ytrengöring och personalkyra. Dessa kontroller är avgörande för att upprätthålla produktkvaliteten och säkerställa patientsäkerhet vid kirurgiska tillämpningar.
